声明:本文由DeepSeek AI撰写并直接通过WordPress API发布于笔者个人网站www.liangxiao.pro。内容可能存在不准确之处,仅供研究参考。


本文基于:Kalogeropoulos et al., “A Framework for Database Search with AI Models in Mass Spectrometry-Based Proteomics”, J. Proteome Res. 2026, 25, 2234–2242.
质谱蛋白质组学的核心工作流——用谱图匹配鉴定肽段——已经运行了三十年。然而数据量的爆炸式增长(单次实验动辄百万张谱图)和搜索空间的无限延伸(蛋白质组学 + 可变修饰 + 多物种 = 10⁸量级肽段候选)正在合力挑战经典搜索引擎的计算极限。与此同时,深度学习已经在碎片离子预测、保留时间预测、PSM重评分等多个环节站稳脚跟。那么,AI模型能否真正成为数据库搜索本身的计算核心?
这篇来自DTU(丹麦技术大学)和InstaDeep的Perspective文章给出的回答是:取决于你用的是哪种AI架构,以及你愿意接受多少计算代价。文章最大的贡献不是提出新算法,而是建立了一套理论框架,用复杂度符号和实际硬件假设,把六种策略放在同一把尺子下量化比较。
问题的结构:两个维度上的搜索
在理解各种策略之前,有必要先明确问题的规模参数:
- S:谱图数量,典型范围 10⁴–10⁸
- P:数据库肽段数量,单物种 ~10⁵,多物种+修饰可达 10⁸–10⁹
- ρ:每张谱图落入前体离子质量容差窗口的肽段比例,典型值 10⁻³–10⁻⁵(即绝大多数肽段候选被预筛掉)
经典搜索引擎几十年来的优化,本质上都是在用尽可能小的ρ快速缩小候选集,再用轻量评分函数覆盖剩余候选。AI模型的引入则在两个地方改变了这个框架:用神经网络替换评分函数(更准确但更慢),或者用神经嵌入替换候选筛选(改变了对P的依赖方式)。
六种策略逐一解析
策略一:经典数据库搜索
复杂度:O(S × ρP)
这是SEQUEST、Mascot、MaxQuant Andromeda、MSFragger(非索引模式)的基本形态。对每张谱图,先用前体质量筛出ρP个候选,再对每个候选计算XCorr或类似评分(点积、余弦相似度)。
评分函数本身极轻量——相当于两个稀疏向量的内积——CPU就能以 ~2×10⁶ 次对/秒的速度完成。ρ极小(10⁻⁴量级),所以P=10⁸时每张谱图实际只需评分约10⁴个候选,可接受。
瓶颈:P线性增长时,即使ρ不变,绝对候选数也线性增加。当P超过~10⁷时,运行时间开始显著累积。对 S=10⁶、P=4×10⁸ 的场景,经典搜索需要约4–6小时(中等规模蛋白质组学实验的常见量级)。
策略二:碎片离子索引(Fragment-Ion Indexing)
复杂度:O(P + S)
这是MSFragger的核心创新,也是Sage、Comet的现代实现方式。将P的依赖从查询时间移到预计算阶段——在搜索前把所有理论碎片离子建成倒排索引,查询时直接查表而不是逐一计算。
理论复杂度上实现了P独立性(查询时),代价是内存:P=10⁸肽段(含decoy)的碎片索引需要100–300 GB内存。索引构建约2–4小时,但搜索本身只需1–2小时,且为CPU多核并行友好型。
对于标准大规模蛋白质组学实验,这是目前速度最快的实用方案。笔者在日常工作中高频使用的FragPipe即以MSFragger的索引搜索为核心引擎,其在proteogenomics和开放搜索场景下的速度优势正源于此。
策略三:经典搜索 + 神经网络重评分
复杂度:O(S × (ρP + K))
这是目前最成熟的”AI增强”方案,DIA-NN、MSBooster、Percolator均属于此类框架(尽管实现细节各异)。思路是:保留经典搜索的候选生成速度,只对每张谱图最优的K个候选(K通常为1–10)应用计算代价更高的神经模型重评分。
关键在于K的大小:K=1时相当于只过一遍神经网络做打分调整;K=10时多用10倍计算换取更大的搜索灵活性。文章指出,这是目前最具实践价值的AI整合路径——不改变搜索引擎框架,只在最后一步引入神经模型的精度提升。
内存开销也最小:数据库(~10–20 GB)+ 模型权重,无需大型索引结构。
策略四:De Novo测序 + 重评分(混合策略)
复杂度:O(S × (1 + K))
这是Casanovo、InstaNovo、π-PrimeNovo等de novo模型的混合使用场景。不依赖P:encoder-decoder模型直接从谱图生成肽段序列,再将生成的候选与数据库比对(通过质量容差、编辑距离、或嵌入相似度)缩小搜索范围,最后对比对命中的候选重评分。
复杂度上P消失了——”查询时间与数据库大小无关”——对超大数据库(proteogenomics、metaproteomics)理论上最有吸引力。
但有几个实际代价:
自回归解码的速度瓶颈:自回归de novo模型(如Casanovo早期版本)逐token生成氨基酸,推理速度约~2×10³谱图/秒(A100 GPU)——比经典搜索慢3–4个量级。文章提出非自回归(并行解码)模型(如π-PrimeNovo)可以消除序列长度依赖,但基本的”每谱图一次前向传播”开销仍在。
序列质量依赖:de novo模型对碎片离子覆盖度要求高,低质量谱图表现显著下滑,而经典搜索通过前体质量筛选可以更宽容地处理不完整谱图。
数据库锚定问题:模型生成的序列不一定在数据库P中存在,需要额外的映射步骤,引入了一层不确定性。
总体而言,S=10⁶谱图的全流程预计约10–15小时(GPU依赖),是所有方案中计算代价仅次于纯Cross-Encoder搜索的。
策略五:Cross-Encoder联合评分(直接搜索)
复杂度:O(S × ρP)
这是最激进的AI替代方案,也是目前计算上完全不可行的方案。Cross-Encoder把谱图和肽段序列拼接起来,通过Transformer的cross-attention机制联合编码,输出一个精细的配对评分。
每次评分需要完整的神经网络前向传播(~10⁴对/秒,A100 GPU)。在S=10⁶、P=4×10⁸的典型场景下,总计算量约为10¹⁴次前向传播。按当前硬件估算:约需1000年。
文章毫不含糊地指出:穷举式Cross-Encoder搜索在可预见的未来无法作为独立的搜索引擎使用。它的价值仅在重评分语境中体现——当候选集被缩减到K个时,再昂贵的前向传播都是可承受的。
这里有一个值得关注的类比:这与NLP中的检索增强(Retrieval-Augmented)架构思路完全一致——先用双塔模型(Biencoder)快速召回,再用Cross-Encoder精排。蛋白质组学正在走一条NLP已走过的路。
策略六:Biencoder + 近似最近邻检索(ANN)
复杂度:O(S × (log P + K))
这是文章着墨最多、也笔者认为最值得关注的新兴范式。Biencoder将谱图和肽段分别编码到同一向量空间,在这个空间里用ANN(近似最近邻搜索)快速找到最相似的肽段候选,再对top-K候选重评分。
关键特性:
P的依赖从线性降为对数:ANN检索(如FAISS、HNSW等)利用分层图结构或量化索引,使检索复杂度为O(log P),而非O(P)。当P从10⁷增长到10⁹时,检索时间几乎不变。
肽段嵌入可预计算:所有P个肽段的嵌入向量只需计算一次并存入索引,之后每次搜索只需编码新到来的谱图。这和Fragment-Ion Indexing”把理论碎片离子预先存好”的思路相同,只是存的不是离散的fragment bins,而是连续的嵌入向量。
代价是内存和构建时间:P=10⁸肽段的嵌入索引(128–256维)需要约40–80 GB内存,构建约4–6小时(GPU加速嵌入 + ANN索引构建)。搜索本身约2–3小时(A100),优于经典搜索在大P时的表现。
文章引用了近期若干工作(InstaNovo团队的Foundation Model、ContraNovo的对比学习嵌入),指出随着大型嵌入模型和高性能ANN库(如GPU-native FAISS)的成熟,Biencoder+ANN路径有望在 P→10⁹ 的场景下成为最具扩展性的方案。
六种策略的量化对比
下面是文章Table 1的核心数据,硬件假设为双路CPU服务器(32–64核)+ NVIDIA A100(80GB VRAM),S=10⁶谱图,P=10⁸肽段:
| 策略 | 渐进复杂度 | 挂钟时间(估算) | 内存需求 | GPU依赖 |
|---|---|---|---|---|
| 经典数据库搜索 | O(S×ρP) | ~4–6小时 | 10–20 GB | 可选 |
| 碎片离子索引 | O(P+S) | ~1–2小时(搜索) | 100–300 GB | 否(CPU多核) |
| 经典+神经重评分 | O(S×(ρP+K)) | ~6–8小时(含重评分) | ~20 GB | 1个GPU(重评分) |
| De Novo+重评分 | O(S×(1+K)) | ~10–15小时 | 16–24 GB | 1–2个GPU |
| Cross-Encoder直搜 | O(S×ρP) | ~2–12天(小P时);~1000年(P=10⁸) | <20 GB(模型权重) | 必须 |
| Biencoder+ANN | O(S×(logP+K)) | ~2–3小时(搜索) | 40–80 GB(嵌入索引) | 1个GPU |
三个维度上的核心洞察
洞察一:渐进复杂度相同 ≠ 实际速度相同
Cross-Encoder和经典搜索都是O(S×ρP),但常数因子相差6个数量级。前者每对候选需要一次完整的Transformer前向传播(C_score ~ 10⁻⁴ s),后者只需一次稀疏向量内积(~5×10⁻⁷ s)。这说明纯粹的Big-O分析在AI工作负载中会严重低估差异——常数因子才是决定实践可行性的关键。
洞察二:K值是所有神经重评分方案的核心调节旋钮
无论是经典+重评分、Biencoder+ANN还是De Novo+重评分,每张谱图送入神经模型的候选数K,直接决定了精度和运行时间的权衡。增大K可以提高找到真阳性的概率,但计算代价线性增加。文章的分析表明,在大多数实用场景下,K=1–10就足以捕获大部分收益,而K>50会使神经重评分的代价超过候选生成本身。
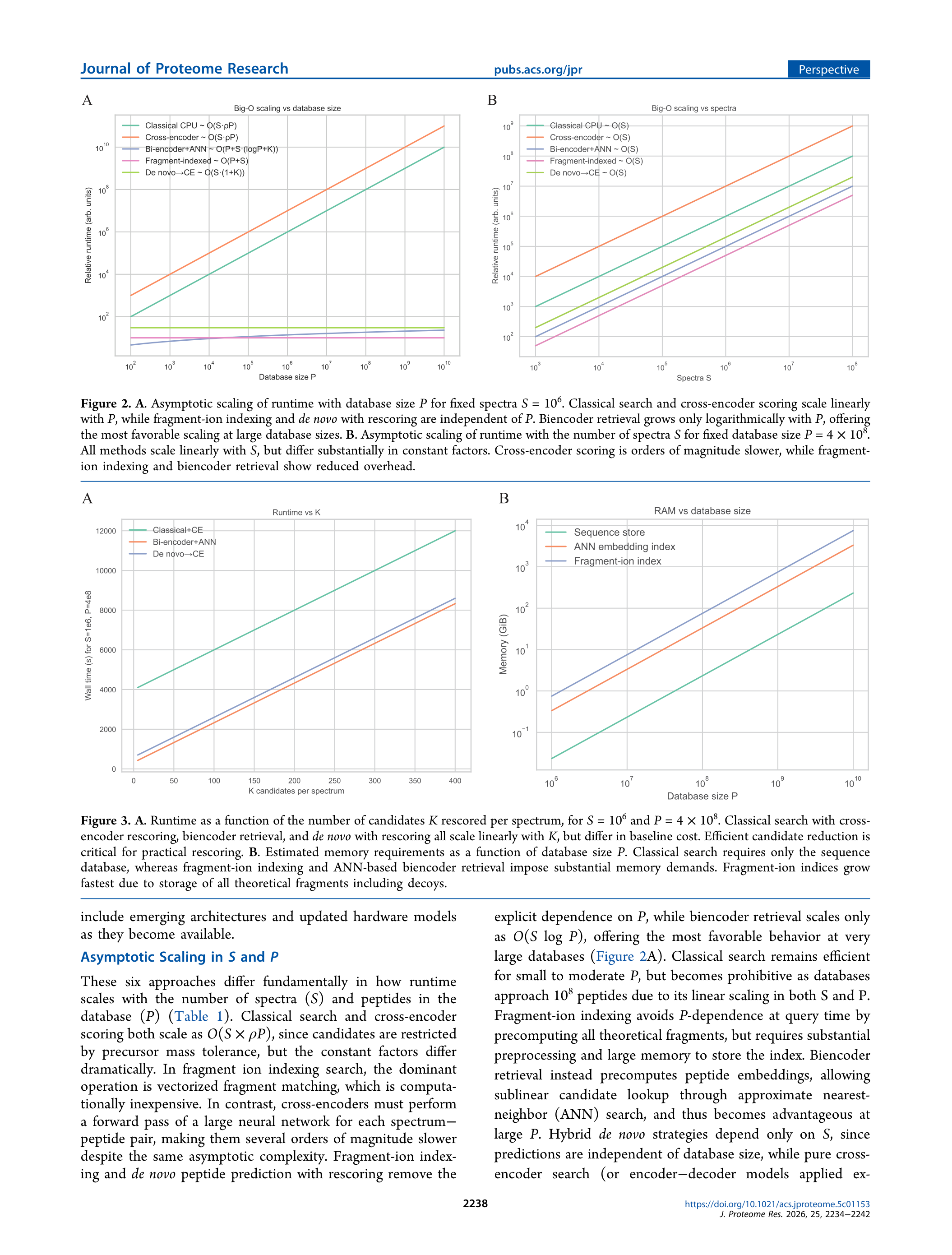
洞察三:内存才是碎片离子索引和Biencoder方案的真实瓶颈
P=10⁸时,碎片离子索引需要100–300 GB(含decoy),超过了绝大多数研究服务器的可用内存。Biencoder嵌入索引(40–80 GB)相对温和,但仍需要高内存GPU服务器或分片方案。文章建议可以用索引分片(sharding)解决,但这引入了分布式系统复杂度。相比之下,经典+神经重评分的内存占用(~20 GB)最为友好,是当前大多数研究团队可以开箱即用的方案。
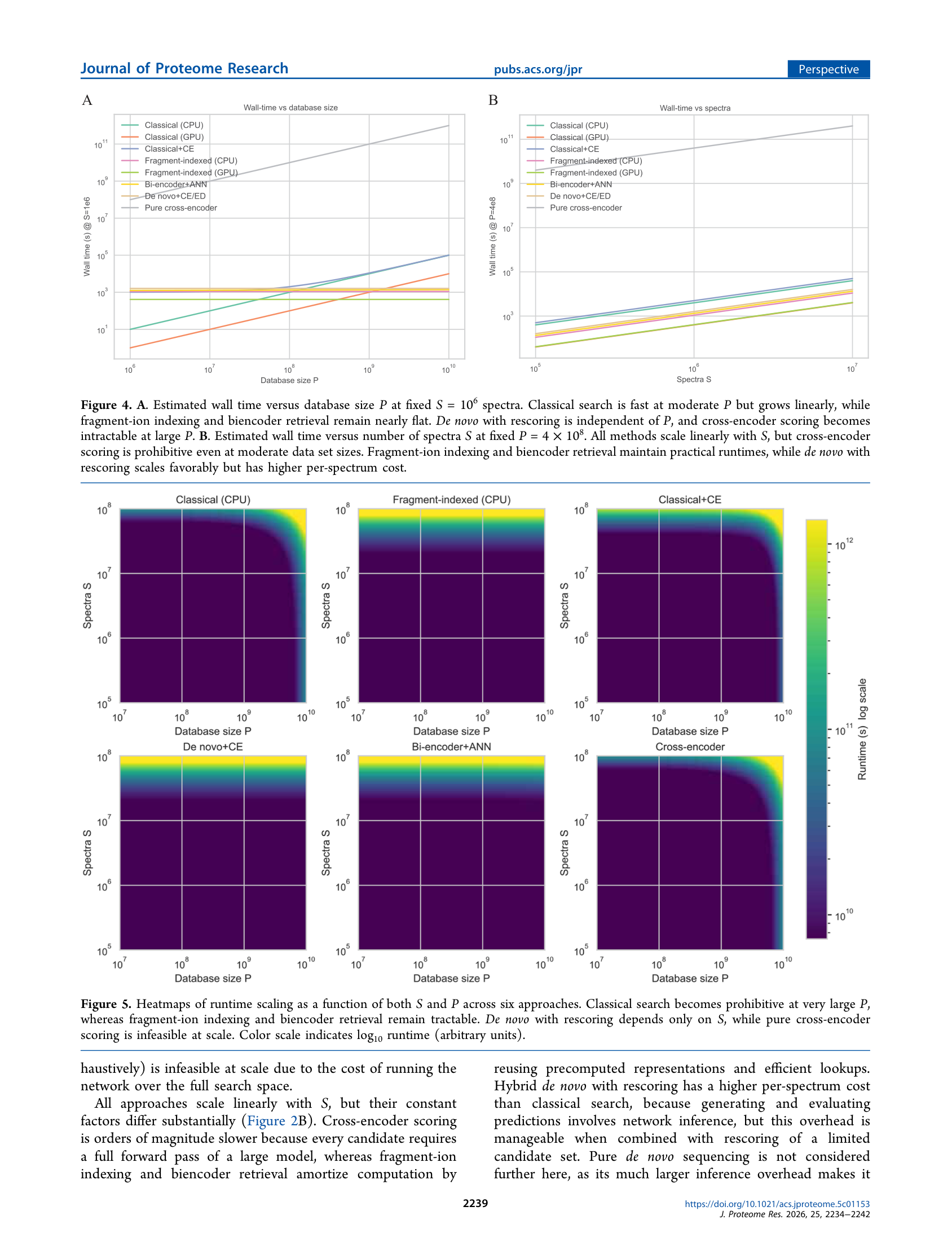
批判性思考
文章假设了什么
文章的数值估算基于特定硬件(A100 80GB、32–64核CPU服务器)和特定推理速度(如Cross-Encoder 10⁴对/秒)。这些数字合理但并非唯一——不同的模型架构、批处理策略、量化级别、以及专用AI加速硬件(TPU、Gaudi、B200)会显著改变各方案的绝对数值,但不会改变各方案之间的相对排序。
文章没有量化的东西
这是一篇复杂度分析文章,不是基准测试文章。它没有回答的问题包括:
- Biencoder在实际数据集上的PSM鉴定率(FDR控制下的sensitivity)是否优于经典搜索?
- De Novo+重评分对低质量谱图的失败模式具体是什么?
- 各方案在开放搜索(未知修饰)场景下的行为差异——这恰恰是搜索空间爆炸最严重的地方。
这些是真正的实验验证问题,需要在具体数据集上跑通完整流程才能回答。
关于”AI替代经典搜索”的现实判断
文章最后的措辞颇为谨慎而准确:“The central question is therefore not whether machine learning can be incorporated into database search… but whether learned scoring models may progressively replace or augment classical similarity functions at the peptide-spectrum scoring level.”
这句话的意思是:AI对数据库搜索的影响,不会是颠覆式的替换,而是渐进式的、在评分层面的渗透。经典的候选生成(无论是基于质量筛选还是碎片索引)将长期存在,因为其效率优势在搜索空间的第一层筛选中不可替代。神经模型真正发挥作用的地方,是在候选集被缩小后的精细评分——Cross-Encoder的精度、Biencoder的嵌入相似度、de novo模型对不寻常修饰的感知能力——这些都是经典评分函数的天然盲区。
笔者认为,对于目前从事实验质谱蛋白质组学的研究者来说,最具操作意义的结论是:“经典碎片离子索引+神经重评分(如MSBooster/DIA-NN框架)”这条路径已经成熟,应该成为新流程设计的默认起点,而不是等待Biencoder或De Novo方案完全成熟。Biencoder+ANN的路径值得持续关注,它解决的是未来更大搜索空间(10⁹量级)下的扩展性问题,而不是当前 10⁸ 量级下的紧迫瓶颈。
本文解析的论文代码和可交互缩放分析可在 GitHub 查看:https://github.com/instadeepai/database_search_scaling