基于质谱的免疫肽组学一直被一系列的技术矛盾束缚:低丰度HLA肽段的检测灵敏度先天瓶颈——这些短肽在细胞内的丰度比常规蛋白质组低了3-4个数量级,且无固定酶切位点,传统富集方法的低回收率让大量功能性肽段始终淹没在背景噪音里;表型与机制的彻底脱节——我们能看到药物让免疫肽组发生了巨大变化,却始终说不清楚这些肽段为什么会变;以及基础研究与临床转化的巨大鸿沟——传统靶点筛选的阴性对照严重不足,导致实验室里的“优质靶点”在临床中频繁出现脱靶毒性,真正落地的寥寥无几。
数天前,Francesca Sacco团队联合Matthias Mann实验室在《iScience》上发表了标题为Kinase inhibition rewires the HLA-I immunopeptidome in Chronic Myeloid Leukemia Targeted therapy reshapes immune visibility in CML的工作(链接:https://doi.org/10.1016/j.isci.2026.115944)。其文章的核心价值在于发现了CML中激酶调控抗原呈递的生物学机制;而笔者更想评述的是其梳理的一套全链条质谱技术体系,将领域内顶尖的单点方法整合为一套闭环的研究工作流,堪称免疫肽组学技术应用的范本。

这项研究从实验设计的起点,就跳出了免疫肽组学领域沿用多年的“盲筛式”逻辑。过往绝大多数“药物对免疫肽组的影响”类研究,本质上都是广撒网的无差别筛选:选几种临床常用的靶向药处理细胞,做完免疫肽组富集后直接找差异肽段,这种模式最大的问题是实验设计完全没有先验的生物学逻辑,最终大概率只能得到一堆零散的差异数据,无法闭环。而这项研究先用ProxPath网络分析完成了靶点预判,基于SIGNOR数据库中已被验证的蛋白因果互作关系,计算了CML病理相关的可成药激酶与抗原加工呈递通路(APPM)的功能连接度,最终锁定了BCR-ABL1、JNK、SFK三个靶点。这确保了后续抑制剂处理带来的免疫肽组变化是与抗原呈递通路有直接功能关联,源头避免了盲筛带来的资源浪费与数据碎片化。
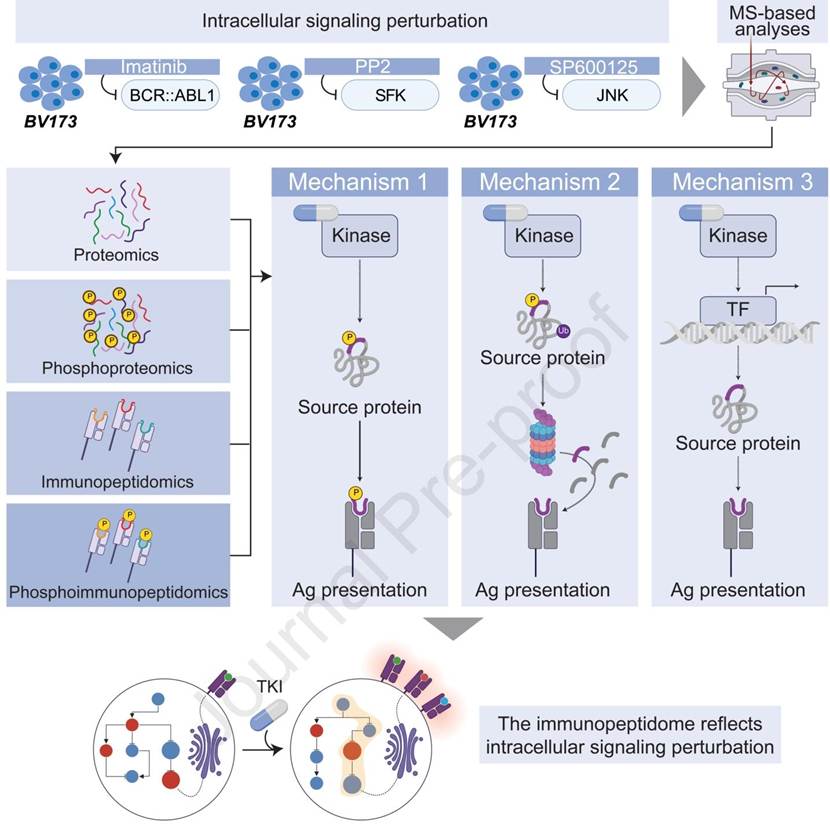
真正决定研究上限的是免疫肽组富集与检测的硬技术实力。这项研究采用的IMBAS-MS工作流是Mann实验室2024年于MCP上发表的新一代免疫肽组学方法,也是目前领域内灵敏度的天花板。IMBAS-MS用生物素化的W6/32抗体+链霉亲和素磁珠的富集方案,生物素与链霉亲和素之间自然界最强的非共价相互作用,既保证了抗体与磁珠的稳定结合,又不会遮蔽抗体的抗原结合位点,最大程度保留了抗体对HLA复合物的捕获效率,同时链霉亲和素磁珠的非特异性结合远低于传统的交联抗体珠子,大幅降低了背景噪音。基于IMBAS(当然,还有明星质谱Bruker timsTOF Ultra2的大功劳;原文配图的Orbitrap有误),研究者基于CML细胞系BV173的生物三重复就鉴定到了21078条唯一的HLA-I配体,且其中超过95%的肽段,都被NetMHCpan工具预测为有结合能力。这为后续所有的差异分析打下了基础。
最后,这项研究通过下表所示的四层多组学的框架,解决了多组学和/或免疫组学领域长期存在的 “知其然,不知其所以然” 的问题。过往多数免疫肽组多组学研究,往往陷入数据简单堆叠的误区,仅通过免疫肽组、转录组、蛋白质组的相关性分析推导机制,无法形成完整的证据链。而这项研究的四层框架,每个维度都精准对应一层调控机制,给出了闭环的证据信息。
| 组学维度 | 核心检测内容 | 关键数据产出 | 解决的核心科学问题 |
| 基础免疫肽组 | HLA-I 结合的全长肽段 | 21078 条唯一配体,4000 + 条受激酶调控的差异肽段 | 哪些抗原肽段的呈递水平发生了变化 |
| 磷酸化免疫肽组 | 结合在 HLA-I 上的磷酸化修饰肽段 | 60 条磷酸化 HLA 肽段,45 条受激酶显著调控 | 肽段本身的翻译后修饰是否直接影响其呈递效率 |
| 全蛋白质组 | 细胞内全蛋白丰度变化 | 定量 7700 + 个蛋白,4700 + 个受激酶显著调控 | 肽段变化是否来自其来源蛋白的整体表达量改变 |
| 磷酸化蛋白质组 | 全蛋白磷酸化位点变化 | 覆盖 17000 + 个磷酸化位点,4400 + 个受激酶显著调控 | 激酶信号如何通过磷酸化修饰,间接调控抗原呈递 |
在这四层框架中,磷酸化免疫肽组的直接检测是技术难度最高的环节。磷酸化修饰的 HLA 肽段是低突变肿瘤的核心潜在免疫治疗靶点,其既来源于自身蛋白,又携带肿瘤异常激酶通路带来的特异性修饰,可弥补低突变肿瘤新生抗原匮乏的短板。但这类肽段的检测存在多重技术壁垒:HLA 结合肽段本身丰度极低,其中磷酸化修饰肽段占比不足 1%,且磷酸化肽段离子化效率远低于非修饰肽段,质谱检测中信噪比极差。正因如此,传统磷酸化免疫肽组研究多采用间接预测的方式:先完成全细胞磷酸化蛋白质组检测,再通过算法预测可被 HLA 结合的磷酸化肽段。这种方法的假阳性率极高,预测结果与体内真实呈递情况往往显著偏差。
这项研究则采用了直接分析的技术路线,直接对富集产物进行磷酸化修饰的质谱检测,最终鉴定到 60 条磷酸化 HLA 肽段,其中 45 条的丰度受激酶抑制剂显著调控,鉴定数量是传统直接检测方法的 3-5 倍。基于这一数据,研究者通过直接质谱证据,首次实验层面建立了 “上游激酶抑制 —— 中间激酶活性变化 —— 下游磷酸化肽段呈递改变” 的完整因果关联。
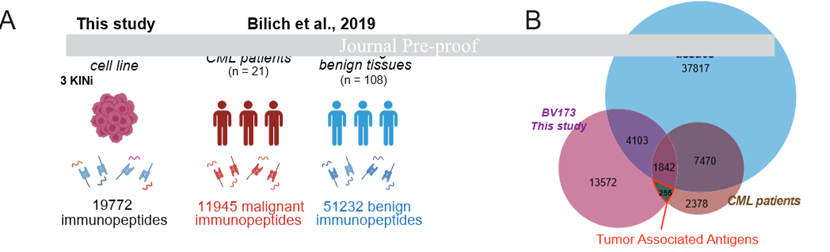
免疫肽组学的核心目标是筛选出可用于临床的安全有效的肿瘤抗原靶点,而靶点筛选的核心,是找到肿瘤中特异性表达、正常组织中不表达的肽段,而非单纯的肿瘤高表达肽段。传统靶点筛选范式的核心缺陷是阴性对照不足,多数研究仅采用 1-2 例正常组织甚至正常细胞系作为对照,无法覆盖人体不同组织、细胞类型的自身免疫肽组全景,易产生大量假阳性靶点。这项研究采用了 “细胞系鉴定 – 原发肿瘤验证 – 大样本正常组织全景排除” 的三级过滤范式,将检测数据集与两个独立大规模参考队列完成三重交叉比对:一是 21 例原发 CML 患者的免疫肽组队列,确保筛选出的肽段真实存在于临床患者肿瘤细胞中,而非细胞系体外培养的假象;二是包含 108 例良性造血组织的 HLA 配体全景图谱,排除正常造血组织中表达的肽段,从源头规避血液系统脱靶风险;三是癌症 – 睾丸抗原(CTA)数据库交叉验证,进一步评估靶点的肿瘤排他性。最终,研究者从 2 万余条初始肽段中,筛选出 255 条仅在 CML 患者样本中出现、良性造血组织中完全检测不到的非突变肿瘤相关抗原(TAA),其中约 50 条高免疫原性 TAA 在激酶抑制剂处理后呈显著上调。
这项工作的技术体系虽完善,但在项目设计和技术新颖度上存在的短板导致其只发表在iScience上。最主要的问题是功能验证缺失。筛选出的核心 TAA 仅完成了免疫原性的生物信息学预测,未通过体外 T 细胞激活实验、体内抗肿瘤模型完成功能验证。此外,研究模型单一。核心实验仅在 BV173 一株急变期 CML 细胞系中完成,未在慢性期 CML 原代患者样本、其他 HLA 亚型细胞中验证调控机制的通用性。然而,尤其是在免疫学领域的读者们大可放心学习和吸收文章给出的技术工作流,用到自己的研究问题上,补短扬长。
Content Refined by AI tools 内容经AI工具修改,审慎阅读