mRNA药物的表达效率高度依赖序列设计,其中5′ UTR选择、密码子优化、UTR-CDS组合对最终蛋白产量影响显著。COVID-19 mRNA疫苗的成功将这一需求推向台前,但序列筛选的现状堪忧:由于多个mRNA变体即便编码完全相同的氨基酸序列(同义密码子替换或UTR替换),其蛋白产物在混合后也无法被ELISA、Western blot或常规定量蛋白质组学区分,每个变体必须单独转染和单独检测,筛选成本随规模线性增长。在抗体工程和基因治疗领域,这一瓶颈尤为突出——当需要比较数十乃至数百个构建体的表达水平时,逐一测试既不经济也不现实。近年来,DNA条码策略在抗体发现和脂质纳米颗粒筛选中已取得显著进展(通过将短DNA序列作为身份标签,在混合筛选后用NGS读出),但这类方法测量的是DNA/RNA的存在而非蛋白终产物本身,难以直接回答”哪个序列表达量最高”这一核心问题。
日立公司Kumano团队(Anal. Chem. 2026, 98, 155-163)在这项工作中扩展了他们此前建立的肽条码策略——将一段约10个氨基酸的肽序列直接融合到目标蛋白C端作为身份标签,共翻译后在蛋白层面标记变体身份,通过肠激酶释放后用LC-MS定量。妙处在于:多个变体可在单次转染中共表达,各自的条码在同一针LC-MS运行中被同时检测,从而将筛选从”逐一”转化为”并行”。该团队此前仅在两个mRNA变体上验证了概念(Rapid Commun. Mass Spectrom. 2024),本研究将规模扩展到九种变体——三种5′ UTR(α-globin、Actb、CD8B来源)与三种eGFP编码序列(野生型、密码子优化、密码子去优化)的全组合,标志着这一方法已初步具备实际筛选价值。
实验设计的核心在于”随机条码分配+事后测序溯源”。文库构建采用Gibson组装一步完成:将线性化骨架质粒、三种UTR片段、三种CDS片段和随机肽条码基因库混合后连接,转化大肠杆菌挑取约500个克隆。条码为10个氨基酸(比此前14-mer缩短,因为14-mer已可检测地影响eGFP表达),其中前6个位置从11种残基(E, P, L, Q, N, A, G, V, Y, W, F)中随机选取——排除了碱性残基以控制总电荷、排除了M/T/S/C/N以避免翻译后修饰和二硫键,C端固定为YRPR(精氨酸增强正模式ESI效率,统一末端抑制电离差异),理论多样性11^6 ≈ 1.8×10^6。转录后的mRNA池通过Nanopore测序(无PCR扩增,避免丰度失真)同时完成两件事:将条码序列归属到对应mRNA变体(通过UTR+CDS序列锚定),以及提供各变体的转录本拷贝数用于后续MS信号归一化。质谱端采用Q Exactive Focus全MS扫描(不做MS/MS),通过±10 ppm质量容差提取EIC后峰面积积分。

条码归属策略是保证数据质量的核心环节。九变体库共鉴定出2,478个不同条码基因,其中583个出现在多个mRNA组中。作者定义”重叠率”为条码在次要mRNA组中的读数 / 在主要组中的读数。规则为:重叠率≥1%的条码完全排除,<1%的则保留在主组中但从次要组排除。此外,同分异构条码(相同m/z但不同序列)也被排除——因为全MS扫描不做MS/MS鉴定。经过双重过滤后,每个mRNA组仍保留至少5个条码用于定量。
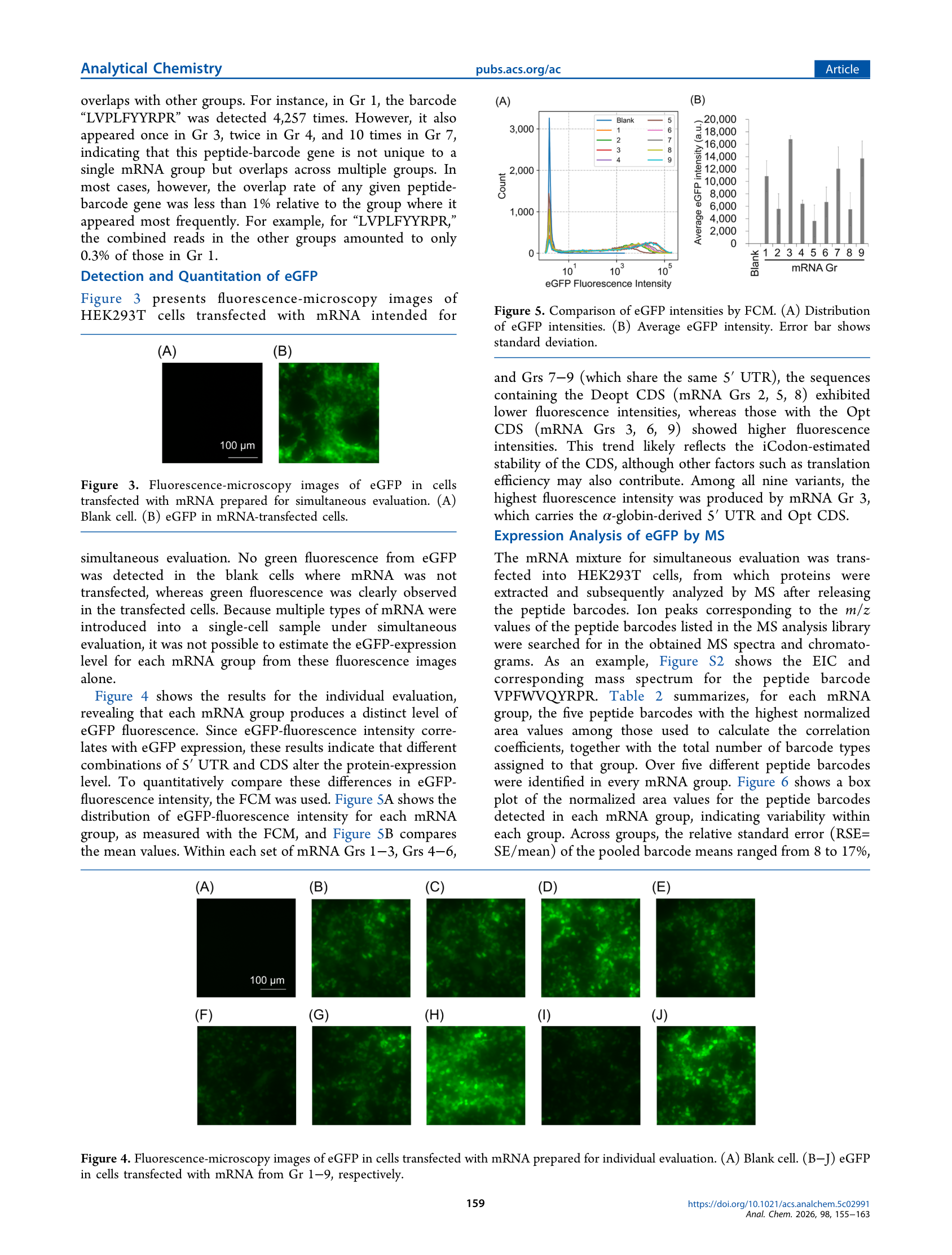
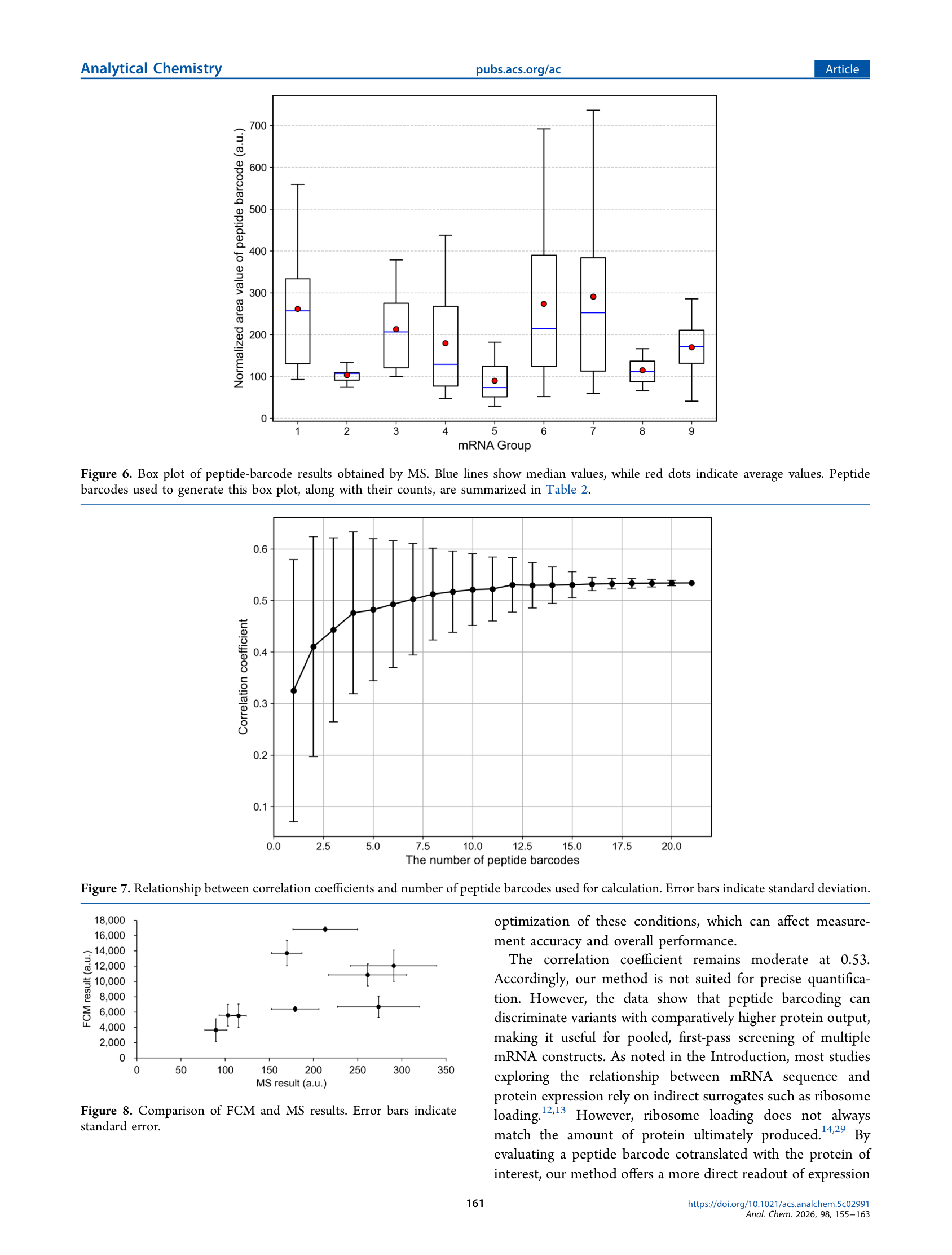
九种变体的独立荧光流式(FCM)结果显示:三种密码子去优化CDS变体荧光最低,三种优化CDS变体最高,α-globin UTR + Opt CDS组合表现最佳,最大差异约5倍。将FCM结果与肽条码LC-MS归一化信号对比,Pearson相关系数随着每组条码数从1增至20后逐步上升至0.53并平台化,误差棒随之收窄;未经Nanopore读数归一化时相关性显著下降;各组归一化条码面积的RSE在8%~17%之间。
在技术层面,有几个设计取舍值得关注。首先是条码长度与蛋白表达影响的根本张力——条码越长越易检测,但对融合蛋白的干扰也越大,这在构象敏感的蛋白结构域中可能是致命问题。其次,全MS扫描与MS/MS的选择体现了灵敏度与序列确认之间的权衡:不碎裂意味着更高duty cycle,但牺牲了区分同分异构体的能力,迫使作者依赖Nanopore测序预排除。这种”测序+质谱”的联合策略与SISCAPA的”免疫富集+质谱”形成对比——后者用抗体捕获降低复杂度,而条码策略用序列唯一性标记身份。
与现有方法的对比则凸显了条码策略的独特定位。DNA条码在抗体和LNP筛选中多作为表型读数(如结合亲和力、递送效率)的代理,而本文将其用于蛋白表达量的直接估计,这是更根本的定量。当前mRNA优化的训练数据来自间接替代指标——mRNA稳定性预测(LinearDesign、iCodon)和核糖体装载(Ribo-seq、MPRA)——但这些替代指标并不总是与蛋白终产量一致(Bicknell et al., Cell Rep. 2024显示降低核糖体负荷反而提高蛋白输出)。肽条码提供的是翻译完成后的蛋白终产物读数,在概念上更接近”地面实况”,但通量远不及MPRA的10^4-10^5量级。与化学标记方法(如TMT)相比,条码策略的独特优势在于标签与蛋白共翻译——不需化学标记反应,不依赖标记效率——其本质上是一种序列编码的样本标识方法。
作者对自身方法的定位十分务实:”不适用于精确定量,但可用于一筛排名。”相关系数0.53的天花板暗示系统误差(条码特异性电离差异、酶切效率差异、色谱行为差异)无法通过简单平均消除;而重叠率是限制扩展的核心瓶颈——随着变体数增加,同一条码出现在多个变体中的概率随之上升。这些边界条件决定了该方法更适合作为筛选漏斗的早期阶段,而非替代独立验证。
值得延伸关注的是日立(Hitachi)公司在生命科学领域的布局。日立集团以工业电子和基础设施著称,但其子公司日立高新技术(Hitachi High-Tech)长期深耕分析仪器领域——电子显微镜、高效液相色谱、质谱系统等产品线在全球科研市场中占有重要份额。本文背后的Kumano团队即来自日立公司内部研发部门(而非高校或研究所),其核心能力正是分析化学方法开发与质谱技术优化。这篇Anal. Chem.论文的意义不仅在于肽条码策略本身——它还折射出一个趋势:分析仪器厂商正在从”卖仪器”走向”输出方法学”,利用自身在分离科学和质谱检测上的深厚积累直接参与生物医药R&D的方法创新。日立在这一赛道的角色介于赛默飞(Thermo Fisher)的”仪器+耗材+服务”全链条模式与安捷伦(Agilent)侧重应用方法开发的”问题导向”路线之间。对于质谱研究者和mRNA药物开发者而言,这类企业实验室产出的方法学论文——虽然有时在生物学深度上不及学术团队,但在质量分析控制的严谨性和可重复性上往往更胜一筹。
内容经AI校正和润色,审慎阅读
论文信息:Kumano et al. “Scaling Peptide-Barcode Approach for Parallel Evaluation of Protein Expression from Multiple mRNA Variants.” Anal. Chem. 2026, 98, 155-163. DOI: 10.1021/acs.analchem.5c02991