声明:本文由Claude AI撰写并直接通过WordPress API发布于笔者个人网站www.liangxiao.pro。内容可能存在不准确之处,仅供研究参考。
基于Genentech与密歇根大学团队发表于 bioRxiv 的预印本论文:Real-time artificial intelligence prediction of peptide characteristics and MSFragger search improves multiplexed quantification of non-canonical HLA presented peptides in clear cell renal cell carcinoma(2026年6月)https://www.biorxiv.org/content/10.64898/2026.05.29.727942v1.full.pdf
一、问题的起点:非经典肽为何难以捕获?
免疫肽组学的核心命题之一,是鉴定HLA分子呈递的肿瘤相关肽段。除了经典蛋白编码区的产物外,大量肽段来源于非经典开放阅读框(nuORF)、长链非编码RNA等曾被视作”非翻译区”的基因座[1][2]。这些非经典肽保留了肿瘤特异性、跨患者共享等诱人的治疗靶点特征,但因为内源性丰度极低,常规的免疫肽组学方法极难对其进行可重复的鉴定和定量。
这里有一个结构性的两难:要想准确定量(尤其是跨样本的TMT多重定量),就需要更长的离子注入时间;而更长的注入时间意味着更少的扫描次数,进一步压缩了本就捉襟见肘的鉴定深度。传统方法在这里陷入了死循环。
二、MIRA-MS的设计逻辑:让质谱在采集时”思考”
Marcu等人提出的MIRA-MS(Model-Informed Real-time Acquisition for Mass Spectrometry),本质上是一个嵌入质谱采集流程的实时决策系统。它的工作逻辑可以概括为三步:
第一步:快速识别。 每个MS²扫描通过Thermo的inSeqAPI接口被实时捕获,由MSFragger-RTS进行碎片离子索引数据库搜索。这里的关键技术突破在于:传统免疫肽组学的数据库搜索是无酶切特异性的(nonspecific),搜索空间巨大——Comet搜索nuORF数据库耗时72ms且常因内存不足而崩溃(需要93GB内存,而仪器标配仅16GB)。MSFragger-RTS通过碎片离子索引将耗时压缩至1ms,并通过远程服务器部署绕过了仪器工作站的内存瓶颈。
第二步:AI验证。 Salud模型(基于Prosit框架但针对ONNX部署优化)对每条候选肽段实时预测碎片谱相似性和保留时间偏移。与离线使用不同,Salud的推理速度极快——碎片谱预测仅需2.7–7.6ms,且已预测过的肽段通过缓存机制降至0.2ms。
第三步:辅助决策。 基于实时FDR过滤和重评分结果,MIRA-MS决定是否为当前肽段触发一次更长时间的定量扫描(SPS-MS3或FTMS2)。简言之,它将昂贵的定量时间精确分配给最值得关注的肽段。
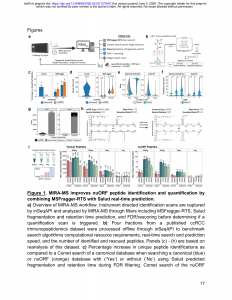
三、核心结果与现实意义
在三个ccRCC细胞系(A-498、TUHR10TKB、VMRC-RCZ)的A*02:01富集样本中,MIRA-MS相比标准FTMS2方法:
- 经典HLA肽定量量提升 45%(+1422条);
- 非经典肽定量量提升 97%(+63条);
- 在pan-HLA-I富集中,提升幅度进一步扩大至 89%(经典)和 107%(非经典)。
更重要的是,实时重评分功能在46个案例中将排名第二的候选肽提升至首位,其中17%的案例纠正了从nuORF到经典序列的错误匹配——这意味着在非经典肽的鉴定中,假阳性问题远比我们想象的严重,而实时AI验证恰好提供了一道内置的质控屏障。
从技术架构看,MIRA-MS代表着一种范式转型:质谱采集不再是”先采集、后分析”的线性流程,而是将数据库搜索、AI预测和FDR控制全部整合进实时采集回路中。这与NeoDiscMS[3]等近年的标签游离方法不同——MIRA-MS保留了TMT多重定量的优势,在实现深度覆盖的同时维持了跨样本的定量可比性。
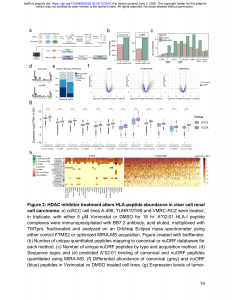
四、难以回避的现实
这篇工作有两个显而易见的特点:其一,核心软件(MSFragger-RTS、inSeqAPI集成、Salud模型)闭源或需要签署商业协议才能获取——MSFragger学术版免费,但商业版由Fragmatics LLC独家授权,且inSeqAPI要求用户签署Thermo的IAPI协议。其二,全文除方法学外全部使用Genentech内部试剂与Thermo Orbitrap Eclipse平台,使得方法在非Thermo生态(如timsTOF或Astral)中的可迁移性尚不明确(笔者注:无法在Astral上使用)。
更为根本的困境在于:国内科研人员往往因各种原因无法获取必要的API接口,这使得MIRA-MS这类依赖实时计算的前沿工具,在实际落地时面临额外的结构性障碍。Genentech的科学家可以在内部高性能服务器上自由部署MSFragger-RTS和Salud推理引擎,但对很多国内实验室而言,光是让Thermo IAPI协议审批通过就可能耗费数月时间、且影响对于昂贵仪器的保修政策。当一个预印本展示了一套优雅的实时AI质谱方案时,全球同行看到的是”技术范式的跃迁”,而不少国内同行的第一反应可能是”我连API都申请不下来”。
五、笔者的观察
MIRA-MS的最重要贡献,不在于它多鉴定了几百条非经典肽——而在于它证明了一个理念:让质谱在采集数据的同时利用AI进行实时决策,是可行的,且效果显著。 这对整个蛋白质组学领域的影响可能远超免疫肽组学本身。
试想:如果实时AI预测可以指导扫描选择,那它同样可以被扩展到来指导PTM位点的靶向定量、指导翻译后修饰异构体的区分、甚至指导单细胞蛋白质组学中极微量样本的智能采集策略。MIRA-MS只是这个方向的第一个成功案例。
当然,笔者也想指出一处批判性观察:论文中将”鉴定到的非经典肽”与”具有免疫原性的治疗靶点”之间画了一个隐含的等号。实际上,在ccRCC模型中鉴定的60条Vorinostat响应性nuORF肽和19条肿瘤特异性nuORF肽中,没有任何一条经过了T细胞免疫原性的实验验证。从”可以被HLA呈递”到”可以被T细胞识别并引发免疫应答”,还有很长一段路。对于临床转化而言,这或许是比鉴定深度更紧迫的问题。
参考文献:
[1]: Ouspenskaia T, et al. Unannotated proteins expand the MHC-I-restricted immunopeptidome in cancer. Nat Biotechnol. 2022;40:209–217. [2]: Chong C, et al. Integrated proteogenomic deep sequencing and analytics accurately identify non-canonical peptides in tumor immunopeptidomes. Nat Commun. 2020;11:1293. [3]: Shapiro IE, et al. Sensitive neoantigen discovery by real-time mutanome-guided immunopeptidomics. Nat Commun. 2025;16:7269.