来自耶鲁大学的Etienne Caron课题组、渥太华大学的Mathieu Lavallée-Adam课题组、以及来自蒙特利尔大学的Julie G. Hussin 协作,日前于Nature Communications上公开了其基于机器学习(笔者注:也包括了深度学习模块)开发的免疫肽组学新算法的工作,软件名称MHCvalidator。论文标题Machine learning-enhanced immunopeptidomics applied to T-cell epitope discovery for COVID-19 vaccines,链接https://doi.org/10.1038/s41467-024-54734-9。

基于质谱的蛋白质组学研究,对于数据的分析基本都需要对谱图匹配数据进行统计验证、以尽可能祛除其中的假阳性匹配。其中最经典的验证算法名为Percolator,通过分析谱图匹配中的“诱饵肽”分布、并基于一套半监督机器学习模型去区分正确匹配和计算后验错误概率及FDR(笔者注:详细资料见http://percolator.ms/)。而本文的特色,就在于开发出了一套免疫肽组学细分领域下的percolator。
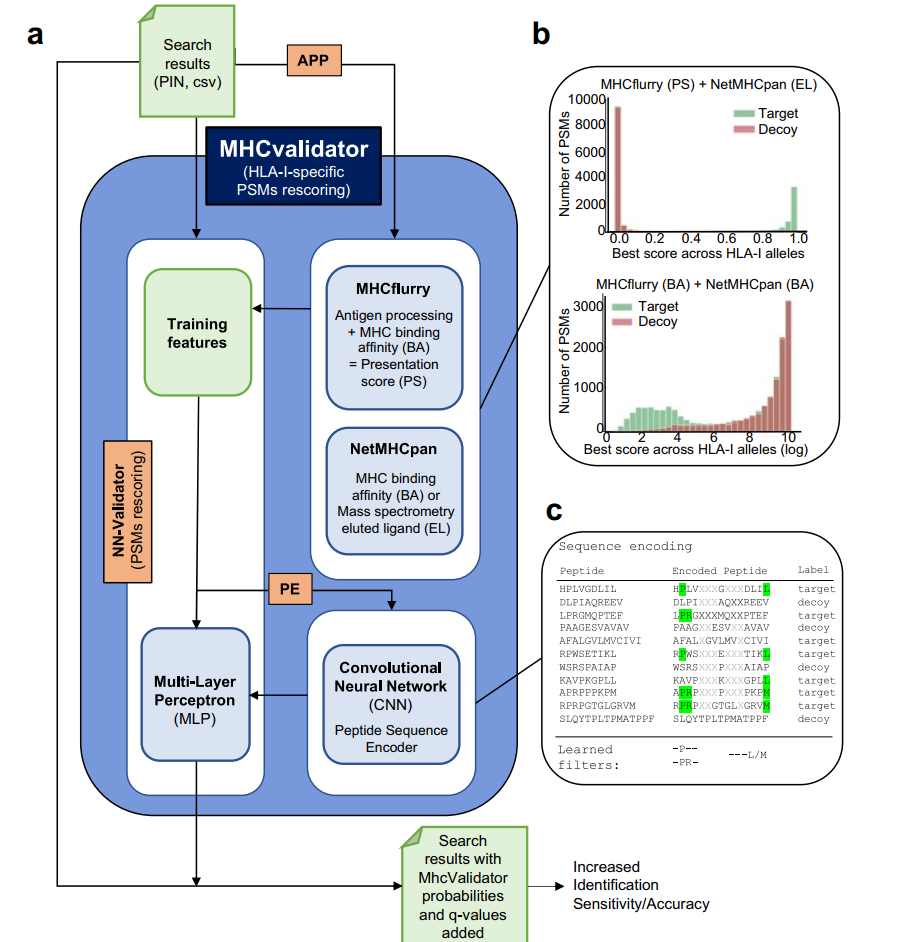
如图所示,MHCvalidator集成了多项新算法,包括MHC肽亲和力预测软件MHCflurry和NetMHCpan用于训练出免疫肽特征、以及CNN神经网络对匹配的谱图进行重打分。因此,获得的结果将多出若干列MHCvalidator整合模型下的打分及概率。
该集成模型对免疫肽组数据分析带来的直接增益包括:
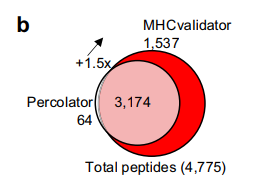
- 在1%肽段水平的FDR下,比Percolator多鉴定出150%的HLA-I肽。
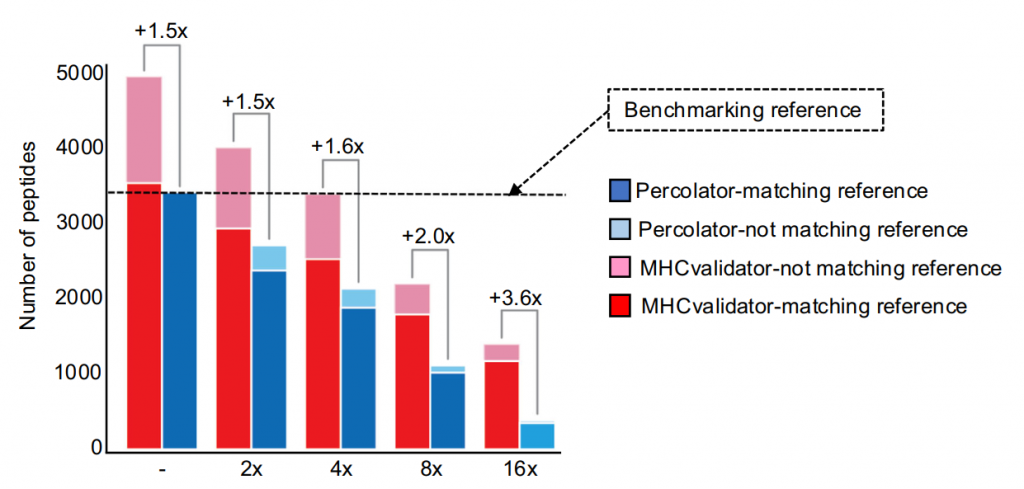
2. 在梯度稀释样本中,MHCvalidator的检测灵敏度较Percolator高出3.6倍。
基于此,研究者们将该模型应用于对SARS-CoV-2感染后的三种细胞系的质谱数据的重分析中。源数据来自于Yardena Samuels等人在2021年公开于Cell Reports的论文,链接https://www.cell.com/cell-reports/fulltext/S2211-1247(21)00681-1。相较于三年前检测出的11条病毒HLA-I肽,使用MHCvalidator后,共24条病毒HLA-I肽可以检测出来,提升了2.2倍;值得注意的是,并不是之前测得的所有肽都在本轮分析中被囊括,如下图。
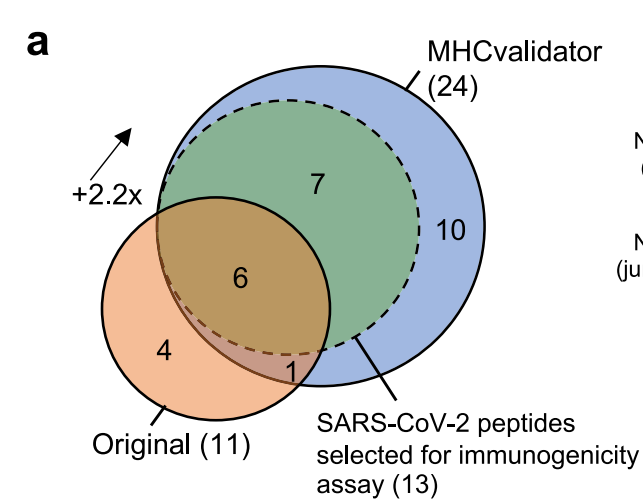
在额外测出的17条多肽中,研究者们验证了其中13条,并发现11条(高达85%)都可以pass ELiSpot实验。包括一个由刺突蛋白+1移码生成的非正典表位LPYPQILLL。研究者们通过调取额外的流行病数据分析证明:约0.8%的COVID-19感染者体内可能存在支持LPYPQILLL生成的基因缺失事件。因此,MHCvalidator还具有检索出因病毒基因突变而产生的非正典表位的能力,这或许就是本文能发表于Nature Communications而非Bioinformatics或MCP等期刊上的重点结果。
文中额外结合EpiTrack等工具分析了疫苗相关表位的地理和时间上的突变动态。
整体来看,这篇论文开发了一套集成的免疫肽组特定的计算框架,不仅能显著提升免疫肽组学数据分析的灵敏度和准确性,还有望推动疫苗表位的高效筛选。笔者感慨,在细分后的领域(包括而不限于各种组学),赛道变多,但起点也在变高。摩拳擦掌的运动员们,有没有想过到达起跑线也得先花些力气呢。