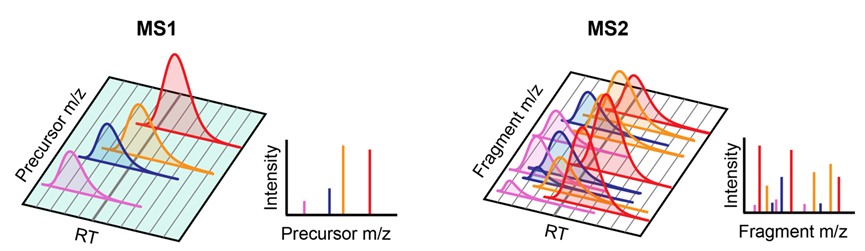
Although DIA-MS is now generally referred to as a SWATH setting that segments the mass range into multiple windows for sequential fragmentation, the original DIA-MS did not look this way. By definition, a scheme is DIA as long as its fragmentation does not rely on signal information from MS1, and the easiest way to achieve this is to fragment the overall inlet ions in the scan range, which we name as full-scan DIA (Figure 1).
The first DIA-MS was developed on a TOF MS and named shotgun collision-induced dissociation (shotgun CID) (1), which is a full-scan DIA setting. In this implementation, the coeluting ions are simultaneously fragmented and recorded, resulting in a maximized duty cycle and highly convoluted MS2 spectra. A variation of shotgun CID, named Parallel CID (p2CID), induces twice-fold fragmentation for the peptides (both in the ion source and in the collision cell) (2), thus increasing the coverage of the detected fragments and their intensities. Full-scan DIA was later implemented on an orbitrap mass analyzer-based MS and named all-ion fragmentation (AIF). This method fragments peptides using advanced higher energy CID (HCD). In addition, it records spectra with a high resolution (100K) (3), generating highly accurate detections with a high dynamic range of peptide intensities (over four orders of magnitude). A congeneric acquisition scheme implemented on the quadruple TOF-MS was named MSE (4, 5), which applies a ramping collision energy for MS2 to increase analysis efficiency.
Full-scan DIA was, at the time, an important innovation. However, it was restricted by two factors: the hardware could not provide accurate and fast sampling, and there was no adequate software to perform automated data analysis. MSE with its continuous development has been applied to biomedical research (6) while the other described methods had fewer applications.
Nevertheless, full-scan DIA bears its own unique advantages. The fast-switching windows do not lose any ions during MS2 and hence in principle enables 100% efficiency of ion usage. This characteristic may also accumulate 10-fold higher data points for an elution profile than SWATH, hence enabling a more accurate quantification and good compatibility with short gradients analysis. With the development of powerful demultiplexing algorithms and AI to extract transition groups as well as to correct interferences, we may see a revival of full-scan DIA in proteomics and peptidomics research.
References
1. Purvine S, Eppel JT, Yi EC, Goodlett DR. Shotgun collision-induced dissociation of peptides using a time of flight mass analyzer. Proteomics. 2003;3(6):847-50.
2. Ramos AA, Yang H, Rosen LE, Yao X. Tandem Parallel Fragmentation of Peptides for Mass Spectrometry. Analytical Chemistry. 2006;78(18):6391-7.
3. Geiger T, Cox J, Mann M. Proteomics on an Orbitrap benchtop mass spectrometer using all-ion fragmentation. Mol Cell Proteomics. 2010;9(10):2252-61.
4. Plumb RS, Johnson KA, Rainville P, Smith BW, Wilson ID, Castro-Perez JM, et al. UPLC/MS(E); a new approach for generating molecular fragment information for biomarker structure elucidation. Rapid Commun Mass Spectrom. 2006;20(13):1989-94.
5. Silva JC, Gorenstein MV, Li G-Z, Vissers JPC, Geromanos SJ. Absolute Quantification of Proteins by LCMSE: A Virtue of Parallel ms Acquisition *S. Molecular & Cellular Proteomics. 2006;5(1):144-56.
6. VC MV, T OA, GH MFS, Gutkoski LC, Cameron LC, M SLF. NanoUPLC-MS(E) reveals differential abundance of gluten proteins in wheat flours of different technological qualities. J Proteomics. 2021;239:104181.